
- “Personalized medicine” is both one of the hottest topics in biomedicine today and one of the oldest concepts in the healing arts. Taking the long view reveals some of the trade-offs in trying to personalize diagnosis and treatment—and suggests that truly personalized medicine will involve not only technological advance, but also moral choices.
It is both one of the hottest topics in biomedicine today and one of the oldest concepts in the healing arts. Visionaries of the genome claim that molecular personalized medicine will eliminate “one size fits all” medicine, which treats the disease, and return us to an older approach, in which the patient was pre-eminent. The revolution, they say, will be “predictive, preventive, personalized, and participatory“—it will be possible to identify why this person has this disease now, and even to prevent disease before it starts. Personalized medicine depends on the individual person. But the individual is not a constant. Over the centuries, the medical individual has evolved along with the increasing reductionism of biomedicine. Medicine has narrowed its scope, moving from the whole person, to part of the body, to proteins, DNA sequences, and single nucleotides. Underlying contemporary, genomic personalized medicine are assumptions that, first, molecular medicine operates on a level that unites us all (indeed, all life), and thus it is the best—even the true—way to explore and describe human individuality. And second, that understanding human individuality on a molecular level will lead willy-nilly to better care and a less alienating medical experience for patients. I think a lot of benefit can come out of the study of genetic constitution and idiosyncrasy—and one can hardly oppose the idea of more personal care. But demonizing one-size-fits-all, promising a revolution, and making fuzzy connections between biochemistry and moral philosophy are risky propositions. Personalized medicine today is backed by money and larded with hype. Setting the medical individual in historical context, we can ask what personalized medicine can and cannot claim. In short, what is the difference between personalized and truly personal?
Seed and soil
The Hippocratic physicians, Aristotle, and Galen all used the concept of diathesis to describe the way a person responds to his environment. They used the term flexibly, to describe everything from a tendency to particular diseases to one’s general constitution or temperament. Around 1800, “diathesis” gained a more specific meaning: it came to signify a constitutional type that made one susceptible to a certain class of diseases. Some diatheses, such as scrofulous, cancerous, or gouty, were believed to be inherited. Others, such as syphilitic, verminous, or gangrenous, were understood as acquired. In its original sense, then, diathesis was related to heredity, but not synonymous with it.

Diathesis and constitution were often discussed in the form of a metaphor of seed and soil. In 1889, the physician Stephen Paget wrote in relation to breast cancer, “When a plant goes to seed, its seeds are carried in all directions; but they can only live and grow if they fall on congenial soil…Certain organs may be ‘predisposed’ to cancer.”[1] The “soil,” he said, could be either a predisposition of certain organs, or diminished resistance. The seed and soil metaphor was also applied to infectious disease. Radical germ theorists such as Robert Koch often argued that the germ was both necessary and sufficient to cause disease. Critics observed that not everyone who was exposed to the germ developed the disease, and that the intensity of the disease often varied. Max von Pettenkofer quaffed a beaker of cholera and suffered only a bit of cramping. The seed-and-soil metaphor helped explain why. In 1894, the great physician William Osler argued:
As a factor in tuberculosis, the soil, then, has a value equal almost to that which relates to the seed, and in taking measures to limit the diffusion of the parasite let us not forget the importance to the possible host of combating inherited weakness, of removing acquired debility, and of maintaining the nutrition at a standard of aggressive activity.[2]
It was a losing battle, though. The germ theorists were winning: diathesis and constitutionalism were already becoming outdated.

“One size fits all” medicine is a direct legacy of the germ theory of disease and of the notion that you can isolate the causative agent in any disease. This was a remarkable advance in medical history. It didn’t matter whether you were a princess or a hack driver, doctors could figure out what you had and make you better. The great legacy therapies of microbial medicine—salvarsan, penicillin, the polio vaccine—represented the first times in medical history that doctors actually cured anyone. One-size-fits-all medicine, then, was positively brilliant, a medical revolution, in an age and culture where infectious disease killed a dominant fraction of the population. But it always had critics, doctors and others who bemoaned the loss of complexity, artistry, humanity from the medical arts. One of those critics was the London physician Archibald Garrod.
The case of the black nappie
Constitution, or soil, had always been associated with heredity; Garrod linked it to genetics. Garrod was a biochemically oriented doctor, interested in the physiological mechanisms of disease.

In 1898, a woman brought her newborn baby to his clinic. It seemed healthy, but she had noticed that its diapers turned an alarming black. The biochemically trained Garrod identified the condition as alkaptonuria, an exceedingly rare and essentially harmless condition believed at the time to be caused by a microbe. Garrod collected all the cases he could, mapped out pedigrees, and published a short note on it, suggesting that the high frequency within the families of his study could hardly be due to chance. The naturalist and evolutionist William Bateson read Garrod’s paper. He had long been interested in variation as the basis of evolution. Bateson had just discovered Gregor Mendel’s work and was emerging as Mendel’s greatest champion in the English language. He saw in Garrod’s alkaptonuria cases powerful ammunition against Mendelism’s critics–proving Mendelian inheritance in man would silence those who argued it a primitive strategy, restricted to plants and lower animals. Bateson and Garrod collaborated to show that alkaptonuria indeed follows a Mendelian recessive pattern. In 1902, Garrod summarized these and other analyses of alkaptonuria as, “Alkaptonuria: a study in chemical individuality.” It was a classic paper, carefully researched, brilliantly argued, compassionate, rational–and widely ignored. Part of the reason for its lack of medical impact was Garrod’s resolutely non-clinical thrust. He was interested not in finding the genes for disease, but in discovering the harmless idiosyncrasies that make us unique. The 1902 paper began to elaborate a biochemical theory of diathesis, which Garrod developed over the succeeding decades. Predisposition to disease, and constitution generally, he said, were biochemical in nature. “Just as no two individuals of a species are absolutely identical in bodily structure,” he wrote, “neither are their chemical processes carried out on exactly the same lines.” He proposed that physiological traits including responses to drugs would be similarly individual and presumably therefore genetic:
The phenomena of obesity and the various tints of hair, skin, and eyes point in the same direction, and if we pass to differences presumably chemical in their basis idiosyncrasies as regards drugs and the various degrees of natural immunity against infections are only less marked in individual human beings and in the several races of mankind than in distinct genera and species of animals.[3]
Obesity, racial features, drug idiosyncrasies, and sensitivity to infectious disease, of course, are now among the primary targets of genetic medicine.
Malaria, drugs, and race
They are also interrelated. For example, take primaquine. After WWI, Germany was cut off from the quinine plantations of Indonesia. German pharmaceutical companies such as Bayer developed synthetic antimalarial drugs, the best of which was primaquine. Primaquine was field-tested in malarial regions such as banana plantations in South America and the Caribbean, especially those run by United Fruit Company. Most of the banana-pickers were black Caribbeans. Researchers found that primaquine was effective, but that about one in ten blacks developed severe anemia when they took it. In whites, this response was extremely rare. This response became known as “primaquine sensitivity.” Today it is recognized as an expression of G6PD deficiency, the most common genetic disease in the world.

During WWII, the Indonesian quinine fields went over to the Germans; now it was the US that needed synthetic anti-malarial drugs. The Army set up research programs in several American prisons—the largest and best-run of these was at Stateville Prison in Illinois. Prisoners were given experimental malaria of different types, and then experimental drugs were tested on them. Racial differences manifested in different roles in the experiments. Ernest Beutler, one of the researchers on the project, said in an interview:
We knew it was only the blacks who were primaquine sensitive. So that was very important. Second place, the blacks didn’t get malaria. They’re resistant to vivax. So we used black prisoners for studies of hemolysis, we used white prisoners usually for malaria.[4]
Thus, skin color became a proxy for susceptibility to malaria. There are other examples, with other drugs, other diseases. Isoniazid was billed as a miracle drug for tuberculosis. But it was soon found that half of all whites and blacks were extremely sensitive to the drug. Physiological studies showed that they metabolized the drug more slowly; their blood drug levels built up quickly, leading to adverse side effects. In such slow acetylators, isoniazid could trigger peripheral neuropathy and even a lupus-like autoimmune reaction. Interestingly, only 15 percent of Asians were slow acetylators. Another drug, succinylcholine, is a muscle relaxant used primarily as a premedication for electroconvulsive therapy. D. R. Gunn and Werner Kalow found that rarely, in one out of 2500 Caucasians, it paralyzes breathing.[5] By the mid-fifties, then, idiosyncratic drug response, susceptibility to infectious disease, and the “various tints of hair, skin, and eyes” were linked in the study of genetic individuality. In 1954, the brilliant and visionary geneticist JBS Haldane could write a small book on biochemistry and genetics. Concluding, he suggested:
The future of biochemical genetics applied to medicine is largely in the study of diatheses and idiosyncrasies, differences of innate make-up which do not necessarily lead to disease, but may do so.[6]
Pharmacogenetics
The young physician Arno Motulsky, of the University of Washington, took that notion as a call to arms. In 1957, he reviewed the literature on drug idiosyncrasy and gave it both context and an audience. “Hereditary, gene-controlled enzymatic factors,” he wrote, “determine why, with identical exposure, certain individuals become ‘sick,’ whereas others are not affected. It is becoming increasingly probable that many of our common diseases depend on genetic-susceptibility determinants of this type.” His short article became a classic and is often cited as the founding paper of pharmacogenetics. The actual term wasn’t coined until two years later, and then in German, by the German researcher Friedrich Vogel: “pharmacogenetik.” In 1962, Werner Kalow published a monograph (in English) on pharmacogenetics. The field soon stalled, however; little was published on the subject through the ‘60s and ‘70s. Pharmacogenetics only really gained traction after the development of gene cloning.[7]
Molecular disease
Molecular approaches to variation had been developing since the late 1940s. In 1949, using the new electrophoresis apparatus—biology’s equivalent of a cyclotron—the great physical chemist Linus Pauling found that the hemoglobin of sickle-cell patients had different mobility than normal hemoglobin. He called sickle cell “a molecular disease.” It was also, of course, considered a “black disease,” for reasons connected to primaquine sensitivity.[8] Pauling once suggested that carriers of sickle cell and other genetic diseases should have their disease status tattooed on their foreheads as a public health measure. It was the kind of step eugenicists of the Progressive Era might have applauded. Pauling was a brilliant and imaginative analyst, but he was not a visionary. He did not foresee that all diseases would become genetic. The study of molecular diseases was greatly aided by technological developments that aided the separation, visualization, and purification of the proteins in biological fluids. Searching for alternative media to replace paper, researchers experimented with glass beads, glass powders, sands, gels, resins, and plant starch. Henry Kunkel used powdered potato starch, available at any grocery. Although inexpensive and compact enough to fit on a tabletop, one could use it to analyze a fairly large sample. Still, electrophoresis with starch grains had its drawbacks. In 1955, Oliver Smithies, working in Toronto, tried cooking the starch grains, so that they formed a gel.

This not only made the medium easier to handle and stain; it made the proteins under study easier to isolate and analyze. Starch gel democratized electrophoresis. Immediately, all sorts of studies emerged characterizing differences in protein mobility; many of these correlated biochemistry with genetic differences. Bateson’s variation had been brought to the molecular level. Sickle cell, the black and molecular disease, continued to play a leading role in the study of genetic idiosyncrasy. In 1957, using both paper electrophoresis and paper chromatography, Vernon Ingram identified the specific amino acid difference between sickle cell hemoglobin and “normal” hemoglobin—specifying Pauling’s “molecular disease.” An idiosyncrasy—or diathesis—now had a specific molecular correlate.[9]
Polymorphism, from proteins to nucleotides
Polymorphism is a population approach to idiosyncrasy. Imported from evolutionary ecology, as a genetic term polymorphism came to connote a regular variation that occurs in at least one percent of the population. Where the ecologist E. B. Ford had studied polymorphisms in moth wing coloration, the physician Harry Harris studied it in human blood proteins. Like Garrod, Harris wanted to know how much non-pathological genetic variation there was in human enzymes. He concluded that polymorphism was likely quite common in humans. Indeed, Harris identified strongly with Garrod; in 1963, he edited a reissue of Garrod’s Croonian Lectures of 1909, Inborn Errors of Metabolism. Together with Lionel Penrose, an English psychologist interested in the genetics of mental disorders, Harris headed up an informal “English school” of Garrodian individualism and biochemical genetics, located at London’s Galton Institute. Harris fulfilled Garrod’s vision by categorizing amino acid polymorphisms and relating them to human biology.
")
Through the ‘50s and ‘60s, young researchers interested in human biochemical genetics streamed through the Galton to learn at the knobby knees of Penrose and Harris. Arno Motulsky penned his 1957 review just after visiting. The Johns Hopkins physician Barton Childs also spent time at the Galton, and later went on to articulate a Garrodian “logic of disease,” based on Garrod’s and Harris’s principles of biochemical individuality. Childs’s vision is now the basis of medical education at Hopkins and elsewhere. Charles Scriver, of McGill University, also studied under Harris and Penrose. He is best known for his work on phenylketonuria, a hereditary metabolic disorder that leads to severe mental retardation, but which is treatable with a low-protein, phenylalanine-free diet. In the 1970s, recombinant DNA and sequencing technologies helped bring polymorphism down to the level of DNA. In 1978, Y. W. Kan and Andreé Dozy returned once again to sickle cell disease, and showed that sickle cell hemoglobin could be distinguished from normal hemoglobin by DNA electrophoresis. The difference can be detected by chopping up the DNA with restriction enzymes, which cut at a characteristic short sequence. The sickle cell mutation disrupts one of those restriction sites, so that the enzyme passes it over, making that fragment longer than normal. Ingram’s single amino acid difference could now be detected by the presence of a particular band on a gel. This became known as a restriction fragment length polymorphism, or RFLP. It was a new way of visualizing polymorphism. In 1980, David Botstein, Ray White, Mark Skolnick, and Ron Davis combined Harry Harris with Kan and Dozy. They realized that the genome must be full of RFLPs. They proposed making a reference map of them, a set of polymorphic mile markers along the chromosomes. “The application of a set of probes for DNA polymorphism to DNA available to us from large pedigrees should provide a new horizon in human genetics,” they wrote grandly. Medical geneticists were beginning to think in terms of databases. Further, Botstein & Co. recognized their method’s potential for medically singling out individuals: “With linkage based on DNA markers, parents whose pedigrees might indicate the possibility of their carrying a deleterious allele could determine prior to pregnancy whether or not they actually carry the allele and, consequently, whether amniocentesis might be necessary.” [10] In other words, whether abortion might be indicated. One should also be able to determine, they continued, whether cancer patients are at risk in advance of symptoms. These are basic principles of personalized or genetic medicine. Further, they wrote, the method would be useful for determining population structure—i.e., identifying racial characteristics by geography and genetics. With RFLP mapping, polymorphism was now divorced from phenotype—it was a purely genetic construct. Researchers then took polymorphism down to the level of single nucleotides. The first single nucleotide polymorphisms, or SNPs, were identified in the late ‘80s—an estimated 1 every 2000 nucleotides. And late in 1998, a database was created to pool all this data. Researchers imagined a high-resolution map of genetic variation—an estimated 10 million variants. It was the ultimate in Garrodian genetic variation. The vision was to use dbSNP to identify any individual’s sensitivities and resiliencies. The end of race? A romantic ideal emerged that the discovery of such enormous variation at the DNA level was not merely a scientific but a social triumph. In 2000, on the announcement of the completion of the draft sequence of the human genome, Craig Venter proclaimed, “the concept of race has no scientific basis.”[11] And NIH director Francis Collins strummed his guitar and sang (to the tune of This Land is Your Land),
We only do this once, it’s our inheritance, Joined by this common thread — black, yellow, white or red, It is our family bond, and now its day has dawned. This draft was made for you and me.[12]

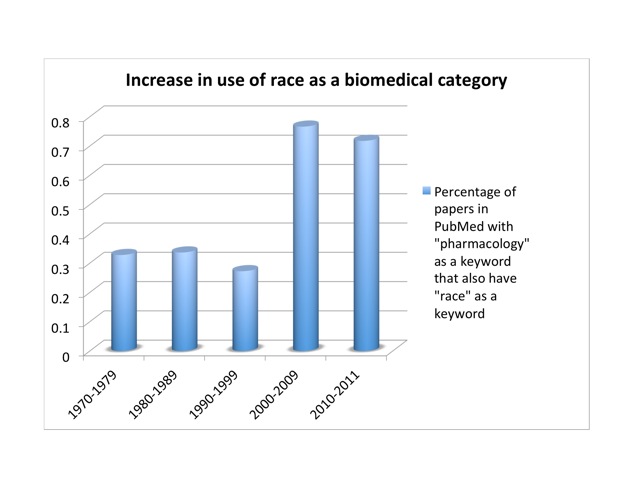
Since then, the genome’s supposed refutation of a biological race concept has become a standard trope among scientists, journalists, and historians.[13] But the SNP database turned out to be too data-rich. Human genetic diversity is far too great to be useful to our poor small brains and computers. Circa 2001, it was discovered that SNPs cluster into groups, or “haplotypes.” Most of the information in a complete SNP map lies in 5% of the SNPs. This brings the number from ten million down to half a million or so. Conveniently, haplotypes cluster by race. In October, 2002, an International HapMap Consortium convened. The HapMap project sampled humanity with 270 people drawn from four populations. There were thirty “trios”—father, mother, and adult child—from the Yoruba of Nigeria—part of an African diaspora widely and condescendingly noted for its literacy. Thirty more trios were white Utahns of European ancestry. Forty-five unrelated individuals were drawn from Japanese in Tokyo and Han Chinese in Beijing (China boasts more than fifty ethnic groups, the largest of which is the Han). No native Australians or Americans, North or South, were included. Conceptually, the HapMap project was a mess. It claimed to explore human diversity while genetically inscribing and condensing racial categories—which in turn were defined by the project in terms of highly cosmopolitan and otherwise problematic groups. It implied that the Yoruba were representative of “Africans,” the Japanese and Chinese of “Asians.” Framed as a corrective to the disastrous but well-intentioned Human Genome Diversity Project of the 1990s, it nevertheless reinforced the racial categories that the genome project was supposed to have shattered. Indeed, Venter’s proclamations and Collins’s corny folksongs notwithstanding, the use of race has actually increased in studies of genetic polymorphism in response to drugs. I looked at the number of papers listed in PubMed that had “pharmacology” as a keyword, and the fraction of those papers that also had “race” as a keyword. That proportion held fairly steady at about a third of a percent from the ‘70s through the ‘90s. But it nearly tripled in the decade after we got the genome, to more than three quarters of a percent: from 34 to 359 publications. Once a sport, a rare mutation in the pharmacological literature, race is now approaching the frequency of a polymorphism. So either race does have a scientific basis after all, or scientists are using a social construct as if it were a biological variable. Either way, there’s a problem.
BiDil and BRCA2 As Beutler used skin color as a proxy for primaquine sensitivity and malaria susceptibility, so physicians today are using it as a proxy for haplotype. For example, take the heart-failure medication BiDil. In 1999, this drug was rejected by the American Food and Drug Administration, because clinical trials did not show sufficient benefit over existing medications. Investigators went back and broke down the data by race. Their study suggested that “therapy for heart failure might appropriately be racially tailored.”[14] The licensing rights were bought by NitroMed, a Boston-area biotech company. Permission was sought and granted to test the drug exclusively in blacks, whose heart failure tends to involve nitric oxide deficiency more often than in people of European descent. On June 23, 2005, FDA approved it for heart failure in black patients. As a result, it became the first drug to be marketed exclusively to blacks. The study’s author claims congestive heart failure is “a different disease” in blacks. This argument thus presupposes that “black” is an objective biological reality, and then identifies health correlates for it. Ethnicity is not so black-and-white. Another well-known case is the gene BRCA2, a polymorphism of which increases the risk of breast cancer. Myriad Genetics, founded by Mark Skolnick, cloned BRCA1 and 2, and took out patents on tests to detect them. Myriad gets a licensing fee for all tests. The BRCA2 mutation is found mainly in Ashkenazi Jews. Due to the wording of the European patent, women being offered the test legally must be asked if they are Ashkenazi-Jewish; if a clinic has not purchased the (quite expensive) license, it can’t administer the test. Gert Matthijs, of the Catholic University of Leuven and head of the patenting and licensing committee of the European Society of Human Genetics, said, “There is something fundamentally wrong if one ethnic group can be singled out by patenting.”[15] The case has been controversial. The patent was challenged, and in 2005, the European Patent Office upheld it. The next year, the EU challenged Myriad. In 2010, an American judge invalidated the Myriad patents. This spring, opening arguments began in the appeal. No one can predict the outcome, but some investors are betting on Myriad. The point is not hypocrisy but internal contradiction. As the ethicist Jonathan Kahn points out, “Biomedical researchers may at once acknowledge concerns about the use of race as a biomedical category, while in practice affirming race as an objective genetic classification.”[16] There’s a deep cognitive dissonance within biomedicine between the public rhetoric and the actual methodology of fields such as pharmacogenetics over the question of whether or not race is real. And this of course has a strong bearing on the question of individuality. Which is it, doctor: are we members of a group, or are we individuals?
Reifying race 
So although biomedicine claims to be moving from “one size fits all” to personalized medicine, in practice, researchers find that race is a necessary intermediate step in getting from the entire population to the individual.
The claim is that individuality is on the horizon—once the databases fill out and testing costs come down, medicine will be truly personalized. In the meantime, though, we’ll put you in a smaller group, which is better than treating everyone the same.
The history shows that treating the individual always involves putting that individual into one or another group. It is neither possible nor even desirable to treat everyone uniquely. When faced with the vastness of human variation, the complexity quickly becomes overwhelming. One has to look for patterns in the data, to group people by their responses. In practice, this often seems to mean typing people according to familiar categories. These categories are of course drawn from the experience of the researchers: if you grow up in a culture where race is real, then those are the categories into which your data fall. Biomedicine is not separate from culture; so long as race exists in our society, it will imprint itself on our science. In this way, the drive for individualism often leads to its opposite: typology. Race becomes reified—it now has an empirical and apparently unbiased basis.
Does personalized equal personal?
Individuality in biomedicine, then, has long been an elusive concept. Biomedical researchers claim with justifiable pride that medicine is beginning to take the individual seriously once again. Specialties such as pharmacogenomics and personalized medicine are increasingly recognizing that not everyone responds the same way to a given disease or a drug. This is a good thing, and could both improve therapeutic effectiveness and reduce incidence of idiosyncratic toxic responses. On the level of technical diagnostics and therapeutics, I see many benefits from tailoring care to whatever extent possible. But that doesn’t make it personal. Science can’t eliminate the concept of race, let alone racial prejudice. It can’t make our doctor take us seriously and treat us respectfully. It’s at best naive and at worst cynical for clinicians and researchers to suggest otherwise. We should always be wary of claims that science & technology will solve social problems. Truly personalized medicine is more than a problem of technology, data collection, and computation. It has to be a moral choice. Acknowledgments This essay is based on a talk delivered to PhD Day in the Division of the Pharmaceutical Sciences, University of Geneva, and was shaped by questions, comments, and discussion afterward. Michiko Kobayashi provided valuable comments and criticisms on both the talk and the essay.
References
Ackerknecht, Erwin. “Diathesis: The Word and the Concept in Medical History.” Bull. Hist. Med. 56 (1982): 317-25.
Bateson, William. “An Address on Mendelian Heredity and Its Application to Man.” British Medical Journal (1906): 61-67.
Bearn, Alexander G. Archibald Garrod and the Individuality of Man. Oxford, U.K.: Clarendon Press, 1993.
Burgio, G. R. “Diathesis and Predisposition: The Evolution of a Concept.” Eur J Pediatr 155, no. 3 (1996): 163-4.
Childs, Barton. “Sir Archibald Garrod’s Conception of Chemical Individuality: A Modern Appreciation.” N Engl J Med 282, no. 2 (1970): 71-77.
Comfort, Nathaniel C. “The Prisoner as Model Organism: Malaria Research at Stateville Penitentiary.” Studies in History and Philosophy of Science, Part C: Studies in History and Philosophy of Biological and Biomedical Sciences 40 (2009): 190-203 (available at academia.edu: http://bit.ly/mjn2CJ)
Comfort, Nathaniel C. “Archibald Edward Garrod.” In Dictionary of Nineteenth-Century British Scientists, edited by Bernard Lightman. London, Chicago: Thoemmes Press/University of Chicago Press, 2004.
Garrod, Archibald Edward. Inborn Errors of Metabolism: The Croonian Lectures Delivered before the Royal College of Physicians of London in June 1908. London: H. Frowde and Hodder & Stoughton, 1909.
———. The Inborn Factors in Disease; an Essay. Oxford: The Clarendon Press, 1931. Hamilton, J. A. “Revitalizing Difference in the Hapmap: Race and Contemporary Human Genetic Variation Research.” The Journal of Law, Medicine & Ethics 36, no. 3 (Fall 2008): 471-7.
Jones, David S., and Roy H. Perlis. “Pharmacogenetics, Race, and Psychiatry: Prospects and Challenges.” Harvard Review of Psychiatry 14, no. 2 (2006): 92-108.
Kay, Lily E. “Laboratory Technology and Biological Knowledge: The Tiselius Electrophoresis Apparatus, 1930-1945.” Hist Philos Life Sci 10, no. 1 (1988): 51-72.
Nicholls, A. G. “What Is a Diathesis?” Canadian Medical Association Journal 18, no. 5 (May 1928): 585-6.
Wailoo, Keith, and Stephen Gregory Pemberton. The Troubled Dream of Genetic Medicine: Ethnicity and Innovation in Tay-Sachs, Cystic Fibrosis, and Sickle Cell Disease. Baltimore: Johns Hopkins University Press, 2006.
Slater, L. B. “Malaria Chemotherapy and the “Kaleidoscopic” Organisation of Biomedical Research During World War II.” [In eng]. Ambix 51, no. 2 (Jul 2004): 107-34.
Snyder, Laurence H. “The Genetic Approach to Human Individuality.” Scientific Monthly 68, no. 3 (1949): 165-71.
Strasser, B. J., and B. Fantini. “Molecular Diseases and Diseased Molecules: Ontological and Epistemological Dimensions.” History and Philosophy of the Life Sciences 20 (1998): 189-214.
Strasser, Bruno J. “Linus Pauling’s “Molecular Diseases”: Between History and Memory.” American Journal of Medical Genetics 115, no. 2 (2002): 83-93.
I would recommend this blog and I got very good information during the course.
Thanks! Spread the word!
thank you
Hello it’s me, I am also visiting this website on a regular basis, this site is truly good and
the visitors are genuinely sharing fastidious thoughts.
Indeed, and welcome! I feel sanctimonious to have such mountebanks as yourself for readers.
Heya just wanted to give you a brief heads up and let you know a few of the images aren’t loading correctly.
I’m not sure why but I think its a linking issue.
I’ve tried it in two different internet browsers and both show the same outcome.
Thank you. That took a few minutes to figure out, but I seem to have fixed it. Everything but the featured image looks ok OMM. Is it all right for you now?